QuantumESPRESSO
License information
Quantum ESPRESSO requires users to register before they can get access to the source code.
Quantum ESPRESSO is coverd by the GNU General Public License. Some information can be found int the "Terms of use" in the user guide and in the "License" file in the Quantum ESPRESSO GitLab repository.
From LUMI/23.09 on, the information is also available in
$EBROOTQUANTUMESPRESSO/share/licenses/QuantumESPRESSO after installation of the package and
loading of the module.
User documentation
Quantum ESPRESSO (QE) is "an integrated suite of Open-Source computer codes for electronic-structure calculations and materials modeling at the nanoscale. It is based on density-functional theory, plane waves, and pseudopotentials.". In general, it runs well on LUMI-C.
There is currently (May 2026 last update) no AMD release of QE that we can support from the LUMI User Support Team. Some releases have support for AMD GPUs via OpenMP offload. However, version 7.4.1omp requires the Cray compiler CCE 15 or older as there are issues with CCE 16 to 18. CCE 15 requires using a ROCm version (5.2, though 5.4 worked also) that is not supported by the current driver on LUMI, and in fact, is even known to fail. Developers are working on a version that works with newer compilers and ROCm™ versions, but mentioned issues with the Cray PE 25.03 and ROCm™ 6.3. We are waiting for further information from them to try with newer compilers and more recent ROCm™ versions, but anything requiring ROCm™ 7 may only come after another system update.
Installing Quantum ESPRESSO
We provide automatic installation scripts for several versions of QE. In general, the installation procedure is described on the EasyBuild page. The step by step procedure to install QE 7.1 is:
- Load the LUMI software environment:
module load LUMI/25.03. - Select the LUMI-C partition:
module load partition/C. - Load the EasyBuild module:
module load EasyBuild-user.
Then, you can run the install command
Builds tend to take 5-10 minutes but depend also on the version of QuantumESPRESSO as the package is expanding in size. When the installation finishes, you will have a module called "QuantumESPRESSO/7.5-cpeGNU-25.03-CPU" installed in your home directory. Load the module to use it
The usual QE binaries, pw.x, ph.x etc. will now be in your PATH. Launch
QE via the Slurm scheduler, e.g. srun pw.x. Please note
that you must do module load LUMI/25.03 partition/C to see your Quantum
Espresso module in the module system. The same applies to the Slurm batch
scripts which you send to the compute nodes.
You can see other versions of QE that can be automatically installed by running the EasyBuild command
or checking the list further down on this page or checking the LUMI-EasyBuild-contrib repository on GitHub directly. It is generally recommended to use the latest version of Quantum Espresso compiled with the most current Cray programming environment. Older version often work, but may not run optimally as the system configuration (software, libraries, drivers, underlying hardware etc.) may have changed since they were originally built.
Example batch scripts
A typical batch job using 2 compute nodes and MPI only:
#!/bin/bash
#SBATCH -J GaAs128
#SBATCH -N 2
#SBATCH --partition=small
#SBATCH -t 00:30:00
#SBATCH --mem=200G
#SBATCH --exclusive --no-requeue
#SBATCH -A project_XYZ
#SBATCH --ntasks-per-node=128
#SBATCH -c 1
export OMP_NUM_THREADS=1
module load LUMI/24.03
module load partition/C
module load QuantumESPRESSO/7.3.1-cpeGNU-24.03
srun pw.x -nk 4 -i gab128.in > gab128.out
A typical batch job with MPI and 4 OpenMP threads per rank:
#!/bin/bash
#SBATCH -J GaAs128
#SBATCH -N 2
#SBATCH --partition=small
#SBATCH -t 00:30:00
#SBATCH --mem=200G
#SBATCH --exclusive --no-requeue
#SBATCH -A project_XYZ
#SBATCH --ntasks-per-node=32
#SBATCH -c 4
export OMP_NUM_THREADS=4
export OMP_PLACES=cores
export OMP_PROC_BIND=close
module load LUMI/24.03
module load partition/C
module load QuantumESPRESSO/7.3.1-cpeGNU-24.03
srun pw.x -nk 4 -i gab128.in > gab128.out
Tuning recommendations
Making use of k-point parallelization (the flag -nk) is very important in
Quantum Espresso. In the following test case, a GaAs supercell with
128-atoms, an SCF cycle completes in a about 1 hour on a LUMI-C
compute node using the default settings without k-point parallelization. By
supplying -nk 2, the runtime is cut to 38 minutes (1.3x faster). K-point
parallelization is so important that it can be advantageous to reduce to the
number of cores used on the compute nodes, and/or increase the number of
k-points just to get even multiples which maximizes the value of -nk.
Consider the case with 14 k-points: 128 cores are not evenly divisible by 14,
which prevents the full use of k-point parallelization. But what about 14*8 =
112 cores? The graph below shows that you can actually gain speed by reducing
the number of active cores and carefully placing the MPI ranks on the right
processor cores.
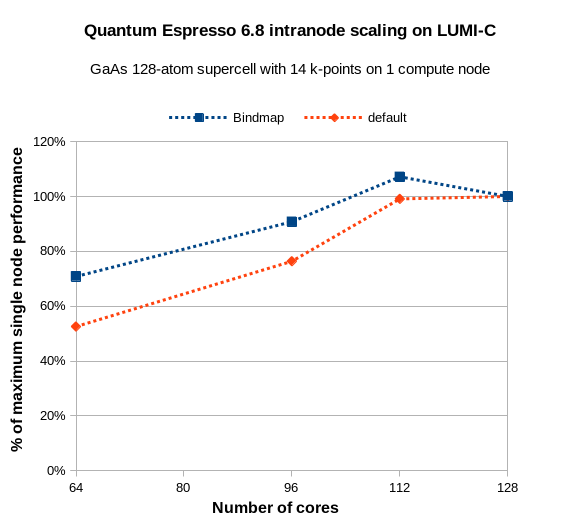
The key to achieving the best performance is to explicitly bind the MPI ranks so that 7 cores are used on each core complex die (CCD). This means that the 8th core needs to be skipped on each CCD in the EPYC CPUs in LUMI-C. There is no easy way to do this in Slurm, other than hard-coding it like this:
$ srun --cpu-bind=map_cpu:0,1,2,3,4,5,6,8,9,10,11,12,13,14,16,17,18,19,20,21,22,24,25,26,27,28,29,30,32,33,34,35,36,37,38,40,41,42,43,44,45,46,48,49,50,51,52,53,54,56,57,58,59,60,61,62,64,65,66,67,68,69,70,72,73,74,75,76,77,78,80,81,82,83,84,85,86,88,89,90,91,92,93,94,96,97,98,99,100,101,102,104,105,106,107,108,109,110,112,113,114,115,116,117,118,120,121,122,123,124,125,126 pw.x ....
It is especially important to do this if you run with e.g. half the number of cores on compute node to free up memory.
OpenMP parallelization can be used primarily to save memory. It is
automatically used when OMP_NUM_THREADS is set to something else than 1 in
the job script. OpenMP seems to work with decent efficiency on LUMI-C. Try
testing lower numbers of OMP_NUM_THREADS first, about 2-4. Typically you will
get about the same speed as with MPI only for regular DFT calculations, maybe
somewhat faster (5-10%). The main benefit, however, is considerably less memory
usage, about 20% less for DFT. We have not tested the effect on exact exchange
or other higher order methods yet.
The default stacksize for OpenMP is small when using the GNU compilers
(system-determined, check with ulimit -s, at the time of writing this is
8MB on LUMI) and this sometimes causes crashes with strange error messages
that point to memory corruption. In that case, you can experiment with
different values for the OMP_STACKSIZE environment variable, and we have seen
the need for values as high as
FFT task parallelization and pencil decomposition are both necessary when
running a large number of cores per pool of k-points. The -ntg flag alone
seems to not work in recent versions of Quantum Espresso. You need to use it
together with pencil decomposition (-pd true), which is an undocumented
feature. Typically, small values of -ntg are sufficient to see a speed-up,
for example -ntg 2 -pd true, but you need to increase -ntg as the number of
cores increases.
For sub-space diagonalization with SCALAPACK (the flag -nd), keeping the
default (maximum) value is typically best. Reducing it will typically just
increase the runtime.
The RMM-DIIS diagonalization algorithm is significantly faster than the
Davidson algorithm on LUMI. This is the option diagonalization=rmm-davidson
in the input file. For regular DFT, it can be 50% faster per SCF step, so it is
worth trying out. As usual, the electronic convergence may not be as a stable
as with the Davidson algorithm, so it depends on the use case whether there is
a net gain.
User-installable modules (and EasyConfigs)
Install with the EasyBuild-user module:
To access module help after installation and get reminded for which stacks and partitions the module is installed, usemodule spider QuantumESPRESSO/<version>.
EasyConfig:
-
EasyConfig QuantumESPRESSO-7.3.1-cpeGNU-24.03.eb, will build QuantumESPRESSO/7.3.1-cpeGNU-24.03
-
EasyConfig QuantumESPRESSO-7.5-cpeGNU-25.03-CPU.eb, will build QuantumESPRESSO/7.5-cpeGNU-25.03-CPU
Technical documentation
-
Quantum ESPRESSO GitLab with direct downloads that don't require registration.
General information
Quantum ESPRESSO is an integrated suite of computer codes for electronic-structure calculations and materials modeling at the nanoscale. It is based on density-functional theory, plane waves, and pseudopotentials (both norm-conserving and ultrasoft).
EasyBuild
Version 6.8.0 for cpeGNU 21.08
- The EasyConfig is derived from the CSCS one
Version 7.0 for cpeGNU 21.12
- The EasyConfig is a port of the 6.8.0 one.
Version 7.1 for cpeGNU 22.06, 22.08
- The EasyConfig is a port of the 7.0 one.
Version 7.2 for cpeGNU 22.12 and 23.09
-
The EasyConfig is a port of the 7.1 one.
-
Added copying of the license information in 23.09
Version 7.3 for cpeGNU 23.09
- Trivial port of the 7.2 EasyConfig file.
Version 7.4.1 for CPE 24.03
-
Port of the 7.3 EasyConfig file, but we ran into issues the EasyBuilders also ran into:
-
There are issues with the d3q version that is pointed to in
external/submodule_commit_hash_records. We used the version used by the EasyBuilders, that has its own issues though for which they have a patch. See also this d3q issue, opened by Davide Grassano who develops for EasyBuild. -
There is an issue with qe-gipaw if the commit is used that
external/submodule_commit_hash_recordspointed to. Code inexternal/qe-gipaw/src/gipaw_setup.f90tries to use the moduleuspp_data, but that module is nowhere to be found in the sources of QE or its dependencies that are installed along with it. There areusppanduspp_paramsmodules, but nouspp_data.It turned out that in the
qe-gipawrepository, a different commit was tagged as version 7.4.1, and we used that commit. -
We also employed another patch from the EasyBuilders that claims to fix an issue with symmetrization.
Archived EasyConfigs
The EasyConfigs below are additional easyconfigs that are not directly available on the system for installation. Users are advised to use the newer ones and these archived ones are unsupported. They are still provided as a source of information should you need this, e.g., to understand the configuration that was used for earlier work on the system.
-
Archived EasyConfigs from LUMI-EasyBuild-contrib - previously user-installable software
-
EasyConfig QuantumESPRESSO-6.8.0-cpeGNU-21.08.eb, with module QuantumESPRESSO/6.8.0-cpeGNU-21.08
-
EasyConfig QuantumESPRESSO-7.0-cpeGNU-21.12.eb, with module QuantumESPRESSO/7.0-cpeGNU-21.12
-
EasyConfig QuantumESPRESSO-7.1-cpeGNU-22.06.eb, with module QuantumESPRESSO/7.1-cpeGNU-22.06
-
EasyConfig QuantumESPRESSO-7.1-cpeGNU-22.08.eb, with module QuantumESPRESSO/7.1-cpeGNU-22.08
-
EasyConfig QuantumESPRESSO-7.2-cpeGNU-22.12.eb, with module QuantumESPRESSO/7.2-cpeGNU-22.12
-
EasyConfig QuantumESPRESSO-7.2-cpeGNU-23.09.eb, with module QuantumESPRESSO/7.2-cpeGNU-23.09
Contains the regular tools, EPW extension and the postprocessing tools. Built with MPI and OpenMP support, and ScaLAPACK.
-
EasyConfig QuantumESPRESSO-7.3-cpeGNU-23.09.eb, with module QuantumESPRESSO/7.3-cpeGNU-23.09
Contains the regular tools, EPW extension and the postprocessing tools. Built with MPI and OpenMP support, and ScaLAPACK.
-